Advanced Genomic Models¶
While standard Transformers (like BERT) are powerful, they have limitations when applied to the vast scale of the genome and the complexity of single-cell data.
In this lecture we introduce two advanced model directions that go beyond a basic SNP encoder:
- Enformer, which is designed for long-range genomic regulation and can connect a variant to distal enhancers far away in the sequence.
- Single-cell foundation models represented by scGPT (with Geneformer as a closely related encoder-style reference), which model how cellular states and gene-expression programs respond across individual cells.
The first direction extends sequence modeling across much longer genomic distances. The second extends foundation modeling from DNA sequence into the cellular transcriptome. Together they show two important ways that genomic AI moves beyond short-window SNP classification.
Long-Range Interactions: Enformer¶
Standard Transformers scale quadratically with sequence length (\(O(N^2)\)). This limits them to windows of ~512-4096 base pairs. However, gene regulation often involves enhancers interacting with promoters over distances of 100,000+ base pairs (100kb).

The Architecture: Dilated Convolutions + Transformer¶
Enformer (DeepMind) solves this by combining two architectures:
- Dilated Convolutions: These layers exponentially increase the "receptive field" (the amount of DNA the model "sees") without exploding the computational cost. They scan the sequence to extract local features.
- Transformer Layers: After the convolutions reduce the sequence length (downsampling), Transformer layers are applied to model the global interactions between these features.
The paper gives a more concrete architecture:
- Input: one-hot encoded DNA of length 196,608 bp (about 197 kb)
- Convolutional stem: 7 convolutional blocks with pooling
- Global context module: 11 transformer blocks
- Cropping layer: trims 320 positions on each side before the prediction head
- Output resolution: 896 bins, where each bin represents 128 bp
- Prediction heads: two organism-specific heads
- human: 5,313 tracks
- mouse: 1,643 tracks
The convolution tower reduces the 196,608-bp input into 1,536 sequence positions, so each latent position roughly corresponds to a 128-bp regulatory chunk. This is a useful compromise: fine enough to represent regulatory elements, but coarse enough to make transformer attention tractable.
Attention Pooling and Relative Position Encoding¶
Two additional design ideas from the paper are worth highlighting:
Attention Pooling¶
Instead of plain max pooling, Enformer uses attention pooling. Within each short pooling window, the model learns which positions are most informative and weights them accordingly. This is more flexible than always taking the maximum value, especially when multiple nearby motifs contribute jointly.
Relative Positional Basis Functions¶
Enformer uses relative positional encodings in the transformer layers rather than relying only on absolute position.
This matters in genomics because the model should distinguish:
- nearby versus far-away regulatory elements
- upstream versus downstream positions relative to a transcription start site
The paper reports that these custom relative positional basis functions improved performance over more standard NLP-style positional encodings.
Why the Cropping Layer Exists¶
The cropping layer is easy to overlook but biologically important. Positions near the far left or far right edge of the input see only one-sided context, whereas positions near the center see context on both sides. Enformer removes these edge positions before computing the loss so the model is trained mainly on positions with a fair, symmetric context window.
Training Tasks and Targets¶
Enformer is trained as a large multitask sequence-to-signal model. It does not predict just one label per sequence. Instead, it predicts many experimental genomic tracks simultaneously.
For the human model, each training example includes:
- 2,131 TF ChIP-seq tracks
- 1,860 histone-mark ChIP-seq tracks
- 684 DNase-seq / ATAC-seq tracks
- 638 CAGE tracks
This is why the final human head has 5,313 outputs.
So the training task is:
- Input: a long DNA sequence
- Output: many position-wise functional genomic signals across many assays and cell types
This is very different from a standard SNP classifier. Enformer first learns a rich map from sequence to regulatory activity, and then SNP effects are computed by comparing predictions for the reference and alternate alleles.
Training Setup in the Paper¶
The paper trains Enformer on both human and mouse genomes in a cross-species multitask setting.
Key details:
- the same Poisson negative log-likelihood loss as Basenji2 is used
- human and mouse batches are alternated during training
- homologous regions are partitioned together into train/validation/test splits
- the model uses sequence augmentation:
- random shifts up to 3 bp
- reverse-complement augmentation
- after joint human-mouse training, the model is fine-tuned on human data
This is a useful teaching point: Enformer is not trained directly on "SNP labels". It is first trained on functional genomics tracks, and only later evaluated on variant-effect tasks.
Downstream Tasks Used in the Paper¶
The paper evaluates Enformer on several biologically important downstream tasks:
Gene Expression Prediction¶
The main benchmark is prediction of CAGE signal at transcription start sites.
The paper reports that Enformer improves mean correlation for gene expression prediction from 0.81 to 0.85 relative to Basenji2, with especially large gains on CAGE tracks. This is important because tissue-specific expression depends strongly on distal enhancers.
Enhancer-Promoter Prioritization¶
The authors use:
- gradient × input
- attention scores
- in silico mutagenesis
to prioritize enhancer-gene pairs validated by CRISPRi experiments. Enformer performs competitively with the ABC model, despite using only DNA sequence as input.
Natural Variant Effect Prediction¶
For GTEx-related evaluation, the paper computes the difference between the reference and alternative allele predictions, sums those differences across the sequence, and uses the resulting feature vector for downstream analysis.
This is tested in two ways:
- genome-wide signed LD profile regression (SLDP) against GTEx summary statistics
- fine-mapped eQTL classification, where likely causal variants are distinguished from matched noncausal variants
The paper reports that Enformer improves the fine-mapped GTEx classifier in 47 of 48 tissues.
Saturation Mutagenesis / MPRA Prediction¶
The paper also evaluates Enformer on massively parallel reporter assay (MPRA) data from CAGI5-style saturation mutagenesis experiments. Again, the model scores a variant by comparing the predicted output for the reference versus alternate allele. Enformer outperforms several competing approaches on average across loci.
Impact on SNP Prediction¶
Enformer can predict gene expression from DNA sequence with remarkable accuracy because it can "see" the distant enhancer that a SNP might disrupt. If a SNP is 50kb away from a gene, a standard BERT model (seeing only 1kb) would miss the connection. Enformer captures it.
In practice, Enformer is especially useful for SNP interpretation when:
- the variant is far from the promoter
- the likely mechanism involves distal enhancers
- you care about signed effects such as whether the alternate allele increases or decreases activity
- you want to score a variant across many regulatory assays, not just one binary label
Paper: Effective gene expression prediction from sequence by integrating long-range interactions
Single-Cell Foundation Models¶
Beyond DNA sequence, we have models trained on Single-Cell RNA-sequencing (scRNA-seq) data. These models learn the "language" of cellular states.
Architectures: scGPT and Geneformer¶
| Feature | scGPT (Generative) | Geneformer (Encoder) |
|---|---|---|
| Input | List of genes and their expression values (ranked). | List of expressed genes (ordered by expression). |
| Tokenization | Each gene is a token. Expression value is an embedding. | Each gene is a token. |
| Objective | Masked Gene Modeling (MGM): Predict expression of masked genes. | Masked Gene Modeling: Predict identity of masked genes. |
| Analogy | Like GPT generating text, it generates a "cell". | Like BERT understanding a sentence (cell). |

scGPT Architecture¶
scGPT (Single-Cell Generative Pre-trained Transformer) treats a single cell as a "sentence" where genes are "words". However, unlike text, the order of genes doesn't matter (permutation invariant), and each gene has a continuous expression value.
The paper pretrains scGPT on 33 million human cells collected from 441 studies across 51 tissues/organs from the CELLxGENE collection. This scale is a major part of the model design: scGPT is intended to learn reusable gene and cell representations, not just task-specific features from one dataset.
Input Embeddings¶
To handle this complexity, scGPT uses a structured input representation. In the paper, the final embedding for each input feature is the element-wise sum of three parts:
- Gene embedding: a learned vector for the gene identity itself, such as TP53 or GATA1.
- Expression embedding: expression values are preprocessed and usually binned before pretraining, then mapped through a value encoder. The paper uses a learnable encoder here so the model can capture the ordinal structure of expression values.
- Condition embedding: per-feature metadata such as perturbation condition, and in downstream tasks also batch or modality information.
The model also prepends a special <cls> token. Its final hidden state is used as the cell embedding, analogous to a sentence-level representation in NLP.
Two practical details from the paper are worth emphasizing:
- the pretraining vocabulary contains the entire human gene set
- to reduce compute, pretraining uses only genes with non-zero expression in each cell
Transformer Backbone & FlashAttention¶
The core is a stack of Transformer blocks with multi-head self-attention. The final hidden states are used in two ways:
- gene-level outputs for expression-prediction objectives
- cell-level output from the
<cls>token for cell-level tasks such as annotation or integration
Because a single cell can contain measurements for thousands to tens of thousands of genes, the attention computation is expensive. The paper therefore uses FlashAttention to make training feasible at these large input sizes.
For integration tasks, scGPT also introduces extra batch tokens and modality tokens. A subtle design choice in the paper is that these batch/modality embeddings are often concatenated to transformer outputs before downstream heads, instead of being treated exactly like ordinary gene tokens throughout the whole transformer. This helps the model account for technical variation without overwhelming the biological attention structure.
Specialized Attention Mask¶
The most distinctive architectural idea in scGPT is its specialized attention mask for non-sequential data.
In language modeling, causal masks work because words have a natural order. In single-cell data, genes do not have a meaningful left-to-right order. The paper therefore splits the input into:
- the
<cls>token - known genes whose expression values are given
- unknown genes whose expression values must be predicted
The masking rule is simple:
- a query token can attend to the
<cls>token - it can attend to all known genes
- an unknown gene cannot attend to other unknown genes
- an unknown gene is still allowed to attend to itself
This gives scGPT a generative workflow while respecting the non-sequential nature of omics data.
Generative Training (Masked Gene Modeling)¶
The paper's pretraining objective is more specific than a generic masked-language-model analogy. scGPT performs generative expression prediction with a regression loss.
During training, a subset of genes is treated as unknown, and the model predicts their expression values with an MLP head using mean squared error.
The paper supports two complementary generation modes:
- gene-prompt generation: predict unknown genes from the known genes in the same cell
- cell-prompt generation: predict gene expression conditioned on the cell representation
These two modes are run consecutively during training, and their losses are added together.
Another important detail is the iterative inference procedure. Instead of predicting every unknown gene in one pass, scGPT predicts a subset, moves the most confident predictions into the known set, and then repeats this for a small number of rounds. This is how the paper mimics autoregressive generation for data that have no natural sequence order.
Fine-Tuning Objectives in the Paper¶
The paper uses several named objectives, depending on the downstream task.
GEP: Gene Expression Prediction¶
This is the basic self-supervised fine-tuning objective. A subset of genes is masked, and the model predicts the masked expression values. It is mainly used to keep learning gene-gene coexpression structure on a new dataset.
GEPC: Gene Expression Prediction for Cell Modeling¶
This objective predicts expression from the cell embedding rather than directly from per-gene hidden states. The goal is to force the <cls> token to become a better summary of the full cell state.
The paper explicitly notes that combining GEP + GEPC works better than using either alone.
ECS: Elastic Cell Similarity¶
This loss shapes the cell embedding space by encouraging already-similar cells to become even more similar, while pushing dissimilar cells farther apart. It is especially helpful for clustering and representation quality.
DAR and DSBN for Batch Correction¶
For integration tasks, scGPT adds explicit anti-batch mechanisms:
- DAR: domain adaptation via reverse backpropagation, where an auxiliary classifier tries to predict sequencing batch from the cell embedding, and gradient reversal pushes the encoder to remove batch-specific signals
- DSBN: domain-specific batch normalization, which keeps separate normalization statistics for different batches
These are used together with expression-modeling objectives when the task requires both biological conservation and batch mixing.
Task-Specific Training Settings¶
The same scGPT backbone is reused across different tasks, but the training setup changes:
- Cell type annotation:
- normalize, log-transform and bin expression values
- keep the common gene set between the pretrained model and the target dataset
- attach a classifier to the cell embedding
- optimize with cross-entropy
- Perturbation response prediction:
- use log1p-transformed continuous values instead of binned values
- append a binary perturbation token at each gene position
- pair a control cell as input with a perturbed cell as target
- use a perturbation-specific GEP objective to predict post-perturbation expression
- scRNA-seq batch integration:
- optimize GEP + GEPC + ECS + DAR + DSBN
- scMultiomic integration:
- reuse pretrained RNA gene embeddings
- train new embeddings for ATAC peaks or proteins
- add modality tokens and batch tokens
- optimize mainly with GEP + GEPC, and DAR if batches are present
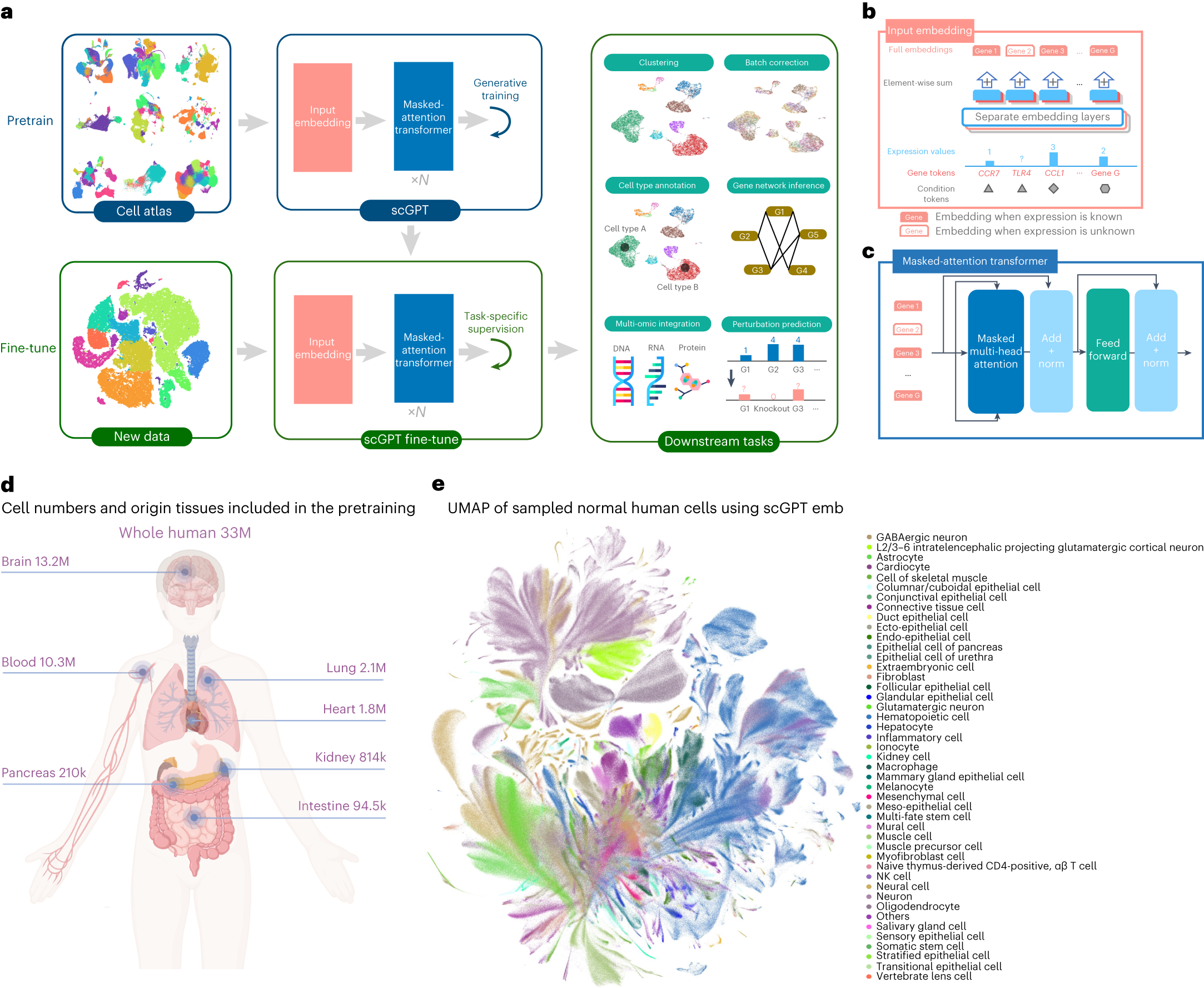 (Source: Cui et al., Nature Methods 2024. The figure shows the input embedding design, the masked-attention transformer, and the pretrain-then-fine-tune workflow across multiple single-cell tasks.)
(Source: Cui et al., Nature Methods 2024. The figure shows the input embedding design, the masked-attention transformer, and the pretrain-then-fine-tune workflow across multiple single-cell tasks.)
Application to SNPs¶
For SNP biology, scGPT is useful because many variants do not act uniformly across all cells. A variant may have a weak bulk effect but a strong cell-type-specific transcriptomic effect.
In practice, you can think of the workflow as:
- a DNA foundation model explains the sequence-side effect of the SNP
- a cell foundation model such as scGPT explains the cell-state response
This lets us ask richer questions such as:
- in which cell types does the SNP have an effect?
- which genes shift expression after the perturbation?
- is the response shared across tissues or highly cell-type specific?
For example, a disease-associated SNP may alter enhancer activity only in immune cells. scGPT helps connect that genotype-side signal to the downstream transcriptome in specific cell populations such as microglia, T cells or hepatocytes.
Papers: * scGPT: scGPT: Towards Building a Foundation Model for Single-Cell Multi-omics Using Generative AI * Geneformer: Transfer learning enables prediction of cell states and phenotypes